
The classical V-model is very often used as the a conceptual model for validation. Most of the V-models are purely designed on a system-based approach, showing in the left wing all sorts of specifications, from URS to design specifications and the right wing all related test phases. Moving away from such a system-based approach in order to follow a risk based approach towards the quality decision making process might impact the existing V-model and a new approach should be used (click picture to enlarge).
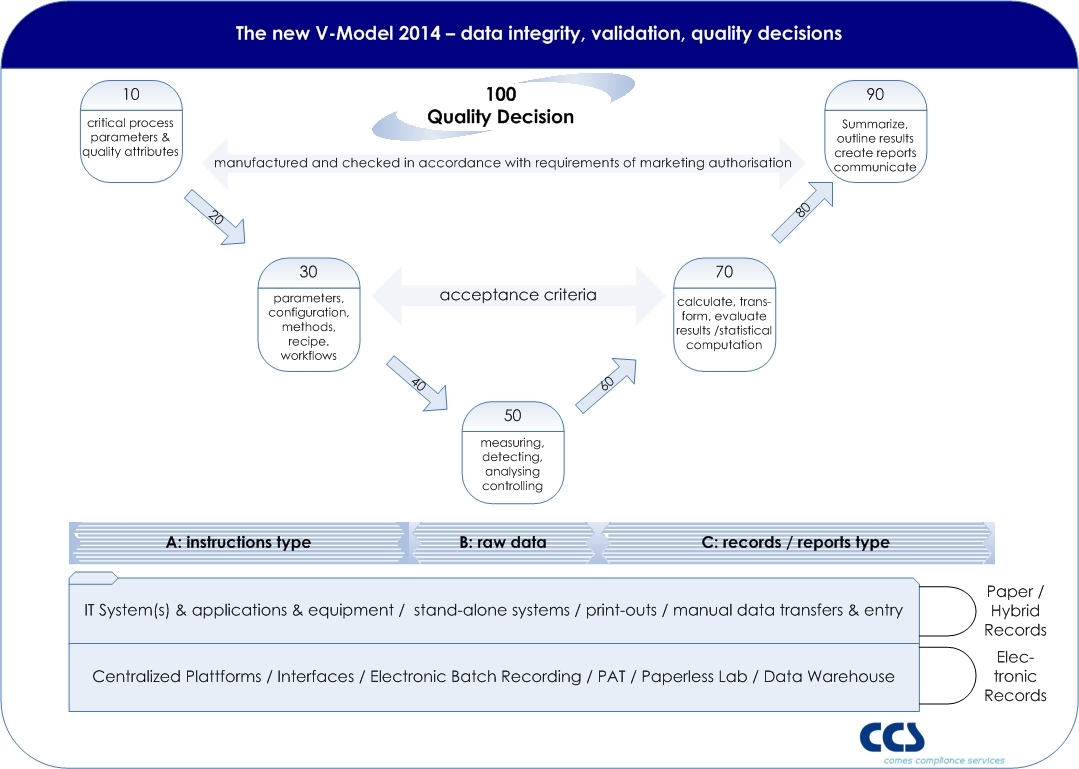
As EU GMP Annex 11 states that applications, and not computerized systems, should be validated, and in real terms we do not validate a computer or system itself, the objective of validation is focused to the quality decision, which is based on the information (recorded in reports and records) derived or given by the entire process, whereas several systems or equipment might be relevant process elements.
EU GMP Chapter 4 – documentation – defines the so called instructions and records/report type. Also the term raw data is used. A GMP record contains such raw data, meta data, master data, on which a decision maker (e.g. Qualified Person, Quality Assurance) is making a quality decision (e.g. Batch Release).
Based on the IT basis such records might support electronic records or is still based on paper records. Ideally all steps (from 10 to 100) are interconnected by the qualified IT architecture.
The fundamental risk is that a wrong quality decision would be made on the basis of insufficient, faulty, tampered or missing data, directly impacting product quality, data integrity, and patient safety. So each system providing data to a GMP relevant record (e.g. Certificate of Analysis, Batch Records, etc.) should be seen as a data sources, forcing, collecting, monitoring, analyzing or calculating data along the entire data flow.
Potentially the highest risk is in each manual step, where data is entered by an operator – whereas a qualified interface of e.g. scanners are reducing the risk of faulty data entries (steps 20, 40, 60, 80).
Step 10 defines critical process parameters, which are derived from product critical attributes, linking product (license) to the process. If a Quality by Design approach is followed, the Design Space will be the basis for the Control Strategy.
A modern approach to computer system validation requires the mapping of the data objects and flows to the final documentation in GMP records. This approach should include the verification of data integrity, as required by EU GMP Annex 11 and US-FDA 21 CFR Part 11.
On the other hand there must be a comprehensive IT strategy including systems, platforms, and interface technology in order to reach the goal of a full electronic recording of all relevant processes. By the way, companies implementing such a holistic data management approach increase efficiency and productivity extensively: Implementing GMP datability & compliance as guarantor of success.
For more information contact us at: talk@comes-services.com or use our online contact form.